These compounds represent new scaffolds for future rhomboid inhibitor and ABP development. In the last decade, small molecule ABPs have substantially impacted protease research, with applications ranging from activity profiling to target discovery and fluorescent imaging. ABPs have also facilitated HTS for ill-characterized enzymes using fluorescent polarization. This HTS has been executed on soluble, but not on membrane enzymes. Recent reports of the first ABPs for intramembrane proteases from the rhomboid family have therefore urged us to investigate FluoPol ABPP for use with membrane enzymes. We have managed this by employing a low concentration of a mild detergent and also found that the surfactant Pluronic F-127 is essential for a good signal-to-noise ratio, probably by facilitating the solubilization of the fluorescent dye. Overall, this resulted in an HTS compatible assay with a high Z-value of 0.9. We are confident that the assay will enable the screening of other poorly characterized membrane-anchored or membrane embedded enzymes. The screening of rhomboids from different organisms is subject of our future research efforts. The special advantage of FluoPol ABPP is that it does not require a substrate, but uses a broad-spectrum ABP. For rhomboids, no small molecule fluorogenic or chromogenic substrates are available as for soluble proteases. One FRET-based polypeptide has been used for screening, but this cannot be used universally. Protein substrates are still the standard assay technique to monitor rhomboid activity. However, the detection of cleavage of these substrates is laborious. Hence, the development and optimization of fluorescent ABPs for rhomboids and other membrane enzymes will likely assist inhibitor discovery for such enzymes. Since the discovery of rhomboids as intramembrane proteases in 2001, inhibitor development has gained momentum slowly. FP-R, for example, reacts with 82% of all mouse metabolic serine hydrolases, which makes it an excellent broad-spectrum ABP. The rhomboid inhibitors based on 4-chloro-isocoumarins have gone through several optimization steps, from the weakly inhibiting DCI, to JLK-6 and S016, which is currently the most potent isocoumarin inhibitor for the E. coli rhomboid GlpG. Still, S016 is more potent against chymotrypsin than against GlpG. The b-lactone scaffold that we have found here, is structurally related to b-lactams. b-lactones are more reactive than b-lactams, and unsurprisingly, b-lactams only act as rhomboid inhibitors when activated with a N-sulfonyl group. The b-lactones 31 and 43 are less potent than the 4chloro-isocoumarin S016, but they have a higher XL-184 potency against GlpG than against trypsin and chymotrypsin. Hence, b-lactones may have the potential to be more selective inhibitors than 4chloro-isocoumarins. Although compounds 31 and 43 also target other ![]() serine hydrolases, the b-lactone scaffold can be readily influenced in its selectivity by changing the substituents on the lactone ring. Compound 43 for example, is an acylated form of 44, the natural product vibralactone. Vibralactone is inactive against rhomboid, probably due to the presence of a polar hydroxyl group that may result in unfavourable interactions with the hydrophobic rhomboid TMDs. When this hydroxyl group is blocked as an ester function in compound 43, it yields an active inhibitor. These structures illustrate the possibility to optimize the b-lactone scaffold for usage against rhomboids. We have shown that the b-lactones covalently and irreversibly react with the active site serine of GlpG. This makes them well suitable for use as ‘warheads’ for ABPs. Compounds 31 and 43 contain an alkyne group in their structure, amenable to click chemistry-mediated derivatization. This feature allowed the direct on-gel visualization of the active rhomboid form. Hence, this study adds two new ABPs to the rhomboid chemical toolbox. Since blactones have already been successfully used for ABPP of serine hydrolases in lysates and live bacterial cells, we expect them to be useful tools for the in vivo functional study of bacterial rhomboids. VE-821 Influenza A viruses infect a wide range of avian and mammalian hosts. The worldwide spread of avian flu as well as the subsequent outbreak of the 2009 H1N1 flu has raised public concerns of the global influenza pandemics due to the high morbidity and mortality.
serine hydrolases, the b-lactone scaffold can be readily influenced in its selectivity by changing the substituents on the lactone ring. Compound 43 for example, is an acylated form of 44, the natural product vibralactone. Vibralactone is inactive against rhomboid, probably due to the presence of a polar hydroxyl group that may result in unfavourable interactions with the hydrophobic rhomboid TMDs. When this hydroxyl group is blocked as an ester function in compound 43, it yields an active inhibitor. These structures illustrate the possibility to optimize the b-lactone scaffold for usage against rhomboids. We have shown that the b-lactones covalently and irreversibly react with the active site serine of GlpG. This makes them well suitable for use as ‘warheads’ for ABPs. Compounds 31 and 43 contain an alkyne group in their structure, amenable to click chemistry-mediated derivatization. This feature allowed the direct on-gel visualization of the active rhomboid form. Hence, this study adds two new ABPs to the rhomboid chemical toolbox. Since blactones have already been successfully used for ABPP of serine hydrolases in lysates and live bacterial cells, we expect them to be useful tools for the in vivo functional study of bacterial rhomboids. VE-821 Influenza A viruses infect a wide range of avian and mammalian hosts. The worldwide spread of avian flu as well as the subsequent outbreak of the 2009 H1N1 flu has raised public concerns of the global influenza pandemics due to the high morbidity and mortality.
Vaccines and antiviral drugs are two available strategies in preventing and controlling influenza virus infections
Leave a reply
.gif) fold of the ICK.
fold of the ICK.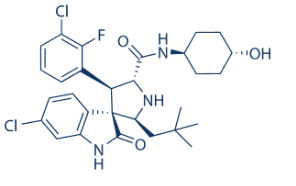 nine was observed when peptide was mixed with liposome vesicles composed of either 1palmitoyl-2-oleoyl-phosphatidylcholine, or a 9:1 molar ratio of POPC and 1-palmitoyl-2-oleoyl-phosphatidylglycerol, indicative of strong binding. Also, addition of DN59 peptide to either POPC or POPC/POPG vesicles containing a fluorescent dye and quencher caused extensive disruption of membrane integrity and leakage of contents to occur at concentrations as low as 2 mM. These observations confirm that DN59 interacts strongly with liposome vesicles and is capable of disrupting artificial lipid bilayers. The observed peptide-lipid membrane interactions are not merely charge based, as binding and disruption occurred with both zwitterionic POPC vesicles as well as negatively-charged 9:1 POPC/POPG vesicles. Supporting these observations, a recent study of the membrane disruption ability of overlapping peptides from dengue virus type 2 C and E proteins showed that E protein stem derived peptides were highly disruptive to liposomes prepared with a wide variety of lipid compositions.
nine was observed when peptide was mixed with liposome vesicles composed of either 1palmitoyl-2-oleoyl-phosphatidylcholine, or a 9:1 molar ratio of POPC and 1-palmitoyl-2-oleoyl-phosphatidylglycerol, indicative of strong binding. Also, addition of DN59 peptide to either POPC or POPC/POPG vesicles containing a fluorescent dye and quencher caused extensive disruption of membrane integrity and leakage of contents to occur at concentrations as low as 2 mM. These observations confirm that DN59 interacts strongly with liposome vesicles and is capable of disrupting artificial lipid bilayers. The observed peptide-lipid membrane interactions are not merely charge based, as binding and disruption occurred with both zwitterionic POPC vesicles as well as negatively-charged 9:1 POPC/POPG vesicles. Supporting these observations, a recent study of the membrane disruption ability of overlapping peptides from dengue virus type 2 C and E proteins showed that E protein stem derived peptides were highly disruptive to liposomes prepared with a wide variety of lipid compositions. phenotypic consequences. Since Insv did not regulate the expression of insb, one possibility is that Insb positively regulates the expression of the insv gene and that Insv antagonizes Notch. Alternatively, the two proteins may act together to repress the expression of Notch target genes via the Su binding sites. Consistent with this, Insv was proposed to repress the expression of Notch target genes by two mechanisms: first in a Su-dependent mechanims, Insv would act as a CSL co-repressor to promote repression through Su binding sites; second, Insv may directly bind DNA via its BEN domain and regulate gene expression in a Su-independent manner. Whether Insb physically interacts with Insv and regulates its transcriptional activities await biochemical studies. While a functional homolog of Insv has recently been characterized in the mouse, no clear homolog of Insb could be easily identified in vertebrates. Thus, deciphering how Insb regulates in flies the activities of Insv and other CSL associated co-repressors, such as H, may provide new insights into molecular mechanisms of co-repression by CSL-associated factors. Finally, while the expression and function of Insb was primarily studied here in the context of sensory organ development, this gene was also expressed at high levels in neuroblasts of the developing larval brain, suggesting that Insb may have a broader role as a Notch antagonist. In conclusion, our study identified Insb as a nuclear SOP/ neuron-specific antagonist of Notch signaling that may act together with Insv to repress the expression of Notch target genes. Assembly of the highly conserved tubulin-like protein FtsZ into a ring structure at the nascent division site initiates the process of cell division in most bacteria. The FtsZ ring serves as a foundation for assembly of the division machinery and constricts at the leading edge of the invaginating septum during cytokinesis. The precise temporal and spatial regulation of cell division is achieved through the actions of a host of proteins, which interact directly with FtsZ to modulate assembly of the cytokinetic ring. Some of these modulators help stabilize FtsZ polymers at midcell and thus maintain the integrity of the cytokinetic ring. In both Bacillus subtilis and Escherichia coli, the location of FtsZ ring formation appears to be dictated in part through the actions of proteins that inhibit FtsZ assembly at aberrant subcellular positions. In B. subtilis, EzrA, a 65 kDa membrane bound protein, plays an important role in both modulatory roles. EzrA is among the first set of proteins to localize to the cytokinetic ring. Null mutations in ezrA reduce the critical concentration of FtsZ required for ring formation in vivo and result in the formation of extra FtsZ rings and septa at cell poles. In contrast to loss of function mutations in other positional regulators of bacterial cell division, the loss of EzrA significantly increases the stability of the medial FtsZ ring, rendering it resistant to overexpression of division inhibitors. Null mutations in ezrA or a point mutation that disrupts EzrA localization to midcell increase cell length by more than 50%, consistent with a model in which EzrA is required for the efficient use of the medial division site.
phenotypic consequences. Since Insv did not regulate the expression of insb, one possibility is that Insb positively regulates the expression of the insv gene and that Insv antagonizes Notch. Alternatively, the two proteins may act together to repress the expression of Notch target genes via the Su binding sites. Consistent with this, Insv was proposed to repress the expression of Notch target genes by two mechanisms: first in a Su-dependent mechanims, Insv would act as a CSL co-repressor to promote repression through Su binding sites; second, Insv may directly bind DNA via its BEN domain and regulate gene expression in a Su-independent manner. Whether Insb physically interacts with Insv and regulates its transcriptional activities await biochemical studies. While a functional homolog of Insv has recently been characterized in the mouse, no clear homolog of Insb could be easily identified in vertebrates. Thus, deciphering how Insb regulates in flies the activities of Insv and other CSL associated co-repressors, such as H, may provide new insights into molecular mechanisms of co-repression by CSL-associated factors. Finally, while the expression and function of Insb was primarily studied here in the context of sensory organ development, this gene was also expressed at high levels in neuroblasts of the developing larval brain, suggesting that Insb may have a broader role as a Notch antagonist. In conclusion, our study identified Insb as a nuclear SOP/ neuron-specific antagonist of Notch signaling that may act together with Insv to repress the expression of Notch target genes. Assembly of the highly conserved tubulin-like protein FtsZ into a ring structure at the nascent division site initiates the process of cell division in most bacteria. The FtsZ ring serves as a foundation for assembly of the division machinery and constricts at the leading edge of the invaginating septum during cytokinesis. The precise temporal and spatial regulation of cell division is achieved through the actions of a host of proteins, which interact directly with FtsZ to modulate assembly of the cytokinetic ring. Some of these modulators help stabilize FtsZ polymers at midcell and thus maintain the integrity of the cytokinetic ring. In both Bacillus subtilis and Escherichia coli, the location of FtsZ ring formation appears to be dictated in part through the actions of proteins that inhibit FtsZ assembly at aberrant subcellular positions. In B. subtilis, EzrA, a 65 kDa membrane bound protein, plays an important role in both modulatory roles. EzrA is among the first set of proteins to localize to the cytokinetic ring. Null mutations in ezrA reduce the critical concentration of FtsZ required for ring formation in vivo and result in the formation of extra FtsZ rings and septa at cell poles. In contrast to loss of function mutations in other positional regulators of bacterial cell division, the loss of EzrA significantly increases the stability of the medial FtsZ ring, rendering it resistant to overexpression of division inhibitors. Null mutations in ezrA or a point mutation that disrupts EzrA localization to midcell increase cell length by more than 50%, consistent with a model in which EzrA is required for the efficient use of the medial division site. and 6P3 when pulling from the loop-closed conformation, contradicting their relative experimental binding affinities. This suggests that the S-site is not the preferred binding site for NHI. The dissociation of FX11, whose binding kept the mobile loop open during conventional MD simulations, turned out to be more difficult than 6P3 when starting from the loop-open conformation. Thus, it appeared that FX11 could bind within the S-site and is indeed a stronger inhibitor than 6P3. Yet, it should be noted that their initial loop conformations are different. The mobile loop in LDHA:FX11S complex is “more closed” than that in LDHA:6P3, and it should be more difficult to unbind FX11 than 6P3 even if they have similar binding affinities within the S-site. The initial loop conformation had a similar impact on the pulling of both dual-site inhibitors. With the mobile loop being initially closed, the pulling of 0SN required more work and larger peak force than that of 1E4, even though 0SN is a slightly weaker inhibitor. Additionally, the work spent on pulling dualsite inhibitors is larger than the combined values of their single-site counterparts, indicating that the linker moiety in both dual-site inhibitors contributes to their binding. The use of a tetrameric model to study LDHA computationally has been attempted previously.However, those studies were based on evidence from either geometry optimization or short-term MD simulations with restraints to prevent large conformational changes.In contrast, the present study employed moderate-length MD simulations with sufficient
and 6P3 when pulling from the loop-closed conformation, contradicting their relative experimental binding affinities. This suggests that the S-site is not the preferred binding site for NHI. The dissociation of FX11, whose binding kept the mobile loop open during conventional MD simulations, turned out to be more difficult than 6P3 when starting from the loop-open conformation. Thus, it appeared that FX11 could bind within the S-site and is indeed a stronger inhibitor than 6P3. Yet, it should be noted that their initial loop conformations are different. The mobile loop in LDHA:FX11S complex is “more closed” than that in LDHA:6P3, and it should be more difficult to unbind FX11 than 6P3 even if they have similar binding affinities within the S-site. The initial loop conformation had a similar impact on the pulling of both dual-site inhibitors. With the mobile loop being initially closed, the pulling of 0SN required more work and larger peak force than that of 1E4, even though 0SN is a slightly weaker inhibitor. Additionally, the work spent on pulling dualsite inhibitors is larger than the combined values of their single-site counterparts, indicating that the linker moiety in both dual-site inhibitors contributes to their binding. The use of a tetrameric model to study LDHA computationally has been attempted previously.However, those studies were based on evidence from either geometry optimization or short-term MD simulations with restraints to prevent large conformational changes.In contrast, the present study employed moderate-length MD simulations with sufficient