Adding biomarker testing as a secondary endpoint to an ongoing phase study represented a timely and . In particular, evidence for a biomarker typically does not KRX-0401 appear early in the drug development process; instead, it usually emerges during phase 2 evaluation and often after a phase 3 study has been initiated. In our case, the PLGF biomarker hypothesis was developed in early-phase testing, with analysis of the phase 2 data occurring while a phase 3 study was ongoing. Consequently, the PLGF hypothesis was added to the phase 3 study following interactions with the FDA. While the option of assessing PLGF as a predictive pharmacodynamic biomarker for motesanib in a larger, independent phase 2 study first represented a scientifically ideal approach, it would have resulted in significant delays in evaluating the hypothesis with no guarantee of a positive outcome. Potentially, a confirmatory prospective run-in design trial may have been considered had the PLGF biomarker hypothesis been confirmed in MONET1. It has been suggested that less than 1% of published cancer biomarkers are routinely used in the clinical setting. Factors identified as contributing to failure to translate biomarkers into the clinic include lack of clinical practicality of the biomarker, hidden biases within the data, an inadequate assay, inappropriate statistical techniques, and lack of biologic plausibility for the biomarker. Although we were not successful in developing a predictive biomarker for motesanib in NSCLC our approach adequately addressed these factors. Biomarker identification was included in early-phase studies, we developed adequate statistical techniques, a robust diagnostic test 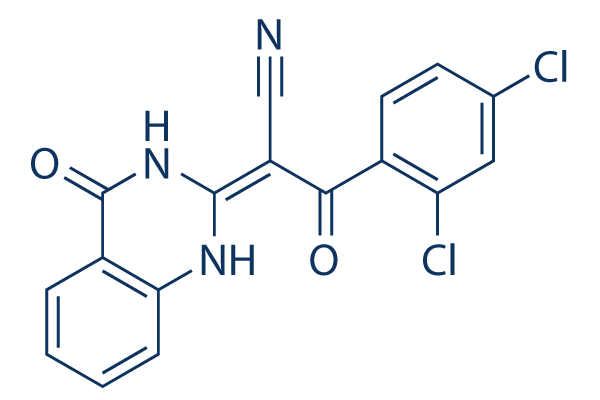 to evaluate PLGF, and engaged early with the US FDA to gain support for our protocol amendment. However, using a pharmacodynamic biomarker as a predictor of efficacy remains an unproven approach. Such biomarkers have typically only been used to identify toxicity issues and there is no precedent that could have guided the development of the biomarker portion of our study. Our experience illustrates several significant challenges to develop predictive pharmacodynamic biomarkers in oncology. Ideal approaches calling for specific study designs and/or sequences of events should be applied wherever possible in an effort to maximize the chances of PB 203580 p38 MAPK inhibitor success; however, they seldom reflect the unpredictable scenarios that may unfold during drug development. Furthermore, a methodical, no-risk approach must be balanced against economic factors and the desire to rapidly identify patient populations that may benefit the most from a potential new treatment. Despite these challenges, it remains important to develop biomarker hypotheses and to subject them to objective evaluation in clinical studies. Development of predictive pharmacodynamic biomarkers remains an opportunity to markedly improve outcomes for patients. With more than 400 millions infections worldwide, malaria remains a major public health issue, principally in sub-Saharan Africa. An effective vaccine would help reduce disease burden, but the best candidates are still in development or evaluation phase. The rapid development of multidrug-resistant Plasmodium parasites necessitates accelerating the discovery of novel antimalarial compounds to meet the needs of the agenda for malaria control and eradication. In humans, Plasmodium sp. development comprises different stages, with the asexual intra�Cerythrocytic forms being responsible for the symptoms of the disease, such as fever, anemia, and cerebral malaria that can lead to death. The erythrocyte invasion by Plasmodium merozoites critically depends on protease activities involved in both the daughter parasites egress from erythrocytes, and invasion into another erythrocyte.
to evaluate PLGF, and engaged early with the US FDA to gain support for our protocol amendment. However, using a pharmacodynamic biomarker as a predictor of efficacy remains an unproven approach. Such biomarkers have typically only been used to identify toxicity issues and there is no precedent that could have guided the development of the biomarker portion of our study. Our experience illustrates several significant challenges to develop predictive pharmacodynamic biomarkers in oncology. Ideal approaches calling for specific study designs and/or sequences of events should be applied wherever possible in an effort to maximize the chances of PB 203580 p38 MAPK inhibitor success; however, they seldom reflect the unpredictable scenarios that may unfold during drug development. Furthermore, a methodical, no-risk approach must be balanced against economic factors and the desire to rapidly identify patient populations that may benefit the most from a potential new treatment. Despite these challenges, it remains important to develop biomarker hypotheses and to subject them to objective evaluation in clinical studies. Development of predictive pharmacodynamic biomarkers remains an opportunity to markedly improve outcomes for patients. With more than 400 millions infections worldwide, malaria remains a major public health issue, principally in sub-Saharan Africa. An effective vaccine would help reduce disease burden, but the best candidates are still in development or evaluation phase. The rapid development of multidrug-resistant Plasmodium parasites necessitates accelerating the discovery of novel antimalarial compounds to meet the needs of the agenda for malaria control and eradication. In humans, Plasmodium sp. development comprises different stages, with the asexual intra�Cerythrocytic forms being responsible for the symptoms of the disease, such as fever, anemia, and cerebral malaria that can lead to death. The erythrocyte invasion by Plasmodium merozoites critically depends on protease activities involved in both the daughter parasites egress from erythrocytes, and invasion into another erythrocyte.
Scientifically robust approach that also illustrates the challenges involved in biomarker development in an oncology setting
Leave a reply
 simulations and MM/GBSA free energy calculations for all possible residues in position P4 and P1 independently, assuming that the effect of the two mutations would be additive. The free energy calculations for the mutants in P4 showed that hydrophobic and bulky residues were preferred for this position. This result fits with the fact that pocket S4 is composed of six hydrophobic residues and seems to have enough space to accommodate larger hydrophobic side-chains than valine. Position P1 instead presents as favorable mutations aromatic residues with polar groups, glutamate and positively charged residues. Surprisingly we found as favorable mutations some positively charged residues, whereas most of sequences recognized by the homologous PfSUB1 present negatively charged or neutral polar sidechains at P1. This might be explained by either the low substrate specificity common to some subtilisins or imprecisions in the structure of the complex.
simulations and MM/GBSA free energy calculations for all possible residues in position P4 and P1 independently, assuming that the effect of the two mutations would be additive. The free energy calculations for the mutants in P4 showed that hydrophobic and bulky residues were preferred for this position. This result fits with the fact that pocket S4 is composed of six hydrophobic residues and seems to have enough space to accommodate larger hydrophobic side-chains than valine. Position P1 instead presents as favorable mutations aromatic residues with polar groups, glutamate and positively charged residues. Surprisingly we found as favorable mutations some positively charged residues, whereas most of sequences recognized by the homologous PfSUB1 present negatively charged or neutral polar sidechains at P1. This might be explained by either the low substrate specificity common to some subtilisins or imprecisions in the structure of the complex.