Its activity was reduced by approximately 50% with an energy load of 30 W, which is 5-fold lower than the energy used for platelet lysis. Using the highenergy Masitinib VEGFR/PDGFR inhibitor sonication protocol employed in previous reported studies, we found that platelet PAI-1 activity was reduced approximately 90%. Taken together, with the activity rates observed in the present study, one would expect sonication to reduce platelet PAI-1 activity to 7�C8%, i.e. to similar levels as reported in previous studies. The magnitude of the reduction in PAI-1 activity was similar when freezing/thawing was used for platelet lysis. However, whereas the reduced activity by sonication was independent of whether tPA was added before or after lysis, the Bortezomib clinical trial underestimation of activity by freezing/thawing could partially be prevented by adding tPA before lysis. Another common procedure for platelet disruption is to use detergents such as Triton X-100. However, it has been shown that Triton X-100 decreases the half-life of active PAI-1 markedly, and 0.2% Triton X-100 decrease the functional half-life of PAI-1 to less than 1 minute at 37uC. Therefore, also with such protocols it is crucial to add tPA before lysis. Since addition of Triton X-100 is not physiological and may facilitate the binding of tPA and PAI-1, we investigated if Triton X-100 affected the results of the Western blot analysis. However, when Triton X-100 was added to the platelets lysed by sonication and freezing/thawing no such enhancement was observed. A potential concern in the present study could have been that the procedure we used in some way could have reactivated PAI-1 although it was in fact inactive in vivo. In vitro PAI-1 can be reactivated by denaturants 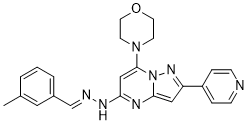 such as SDS, guanidine HCl, and urea, and it has also been suggested that negatively charged phospholipids exposed on the surface of activated platelets could reactivate PAI-1. On the other hand, it has been reported that SDS may cause dissociation of the tPA-PAI-1 complex. To rule out the possibility that our results were due to reactivation and/or dissociation of the tPA-PAI-1 complex formed, we performed a series of experiments both with and without SDS in the loading buffer before electrophoresis. However, these studies showed no detectable differences in PAI-1 activity whether SDS was present or not. This is in agreement with a previous study reporting that the SDS-activatable form of PAI-1 might not be present in human platelets. How, then, could the activity of PAI-1 be preserved for such a prolonged period of time in the platelet? A potential mechanism has been suggested by Lang and Schleef, who showed that platelets possess a unique mechanism for stabilization of active PAI-1, by packaging together with other large a-granule proteins in a calcium-dependent manner. Active PAI-1 in plasma is stabilized by binding to vitronectin which has also been detected in platelet a-granules. However, some studies have failed to detect the vitronectin-PAI-1 complex in platelets and it is therefore controversial whether vitronectin is the stabilizing factor of PAI-1 in platelets. This issue remains to be evaluated. From a clinical perspective, there is compelling evidence that platelet-derived PAI-1 has an important physiologic and pathophysiologic role in making platelet-rich blood clots resistant to both endogenous and pharmacological thrombolysis. Despite this, most previous studies have reported activity levels of platelet PAI-1 that are probably far too low to explain its putative functional role.
such as SDS, guanidine HCl, and urea, and it has also been suggested that negatively charged phospholipids exposed on the surface of activated platelets could reactivate PAI-1. On the other hand, it has been reported that SDS may cause dissociation of the tPA-PAI-1 complex. To rule out the possibility that our results were due to reactivation and/or dissociation of the tPA-PAI-1 complex formed, we performed a series of experiments both with and without SDS in the loading buffer before electrophoresis. However, these studies showed no detectable differences in PAI-1 activity whether SDS was present or not. This is in agreement with a previous study reporting that the SDS-activatable form of PAI-1 might not be present in human platelets. How, then, could the activity of PAI-1 be preserved for such a prolonged period of time in the platelet? A potential mechanism has been suggested by Lang and Schleef, who showed that platelets possess a unique mechanism for stabilization of active PAI-1, by packaging together with other large a-granule proteins in a calcium-dependent manner. Active PAI-1 in plasma is stabilized by binding to vitronectin which has also been detected in platelet a-granules. However, some studies have failed to detect the vitronectin-PAI-1 complex in platelets and it is therefore controversial whether vitronectin is the stabilizing factor of PAI-1 in platelets. This issue remains to be evaluated. From a clinical perspective, there is compelling evidence that platelet-derived PAI-1 has an important physiologic and pathophysiologic role in making platelet-rich blood clots resistant to both endogenous and pharmacological thrombolysis. Despite this, most previous studies have reported activity levels of platelet PAI-1 that are probably far too low to explain its putative functional role.
Our results may provide the missing clue to reconcile the seemingly contradictory findings
Leave a reply
 proliferation, survival and migration. VEGF and VEGFR are also expressed by some tumor cells, like MM, acting in a functional autocrine loop capable of directly stimulating the growth and survival of MM cells. In this study, we have shown lovastatin does indeed inhibit ligand-induced VEGFR-2 activation through inhibition of receptor internalization resulting in diminished AKT activation in HUVEC and MM cells. Lovastatin treatment re-organized the actin cytoskeleton, inhibited proliferation and induced apoptosis of HUVEC at therapeutically relevant doses despite addition of exogenous VEGF. AKT activation, which mediates cell survival, along with its downstream targets S6K1 and 4EBP1 were significantly inhibited by lovastatin treatment. Combining lovastatin with VEGFR-TKIs also induced synergistic cytotoxicity of HUVEC cells. Due to their role in promoting tumor neovascularization, inhibiting the function of VEGF and VEGFR has been the focus of a number of therapeutic approaches. The limited clinical responses associated with these agents have been associated with their ability to promote disease stabilization and rarely induce tumor regression. Thus, agents that can cooperate and enhance the activity of VEGFR-TKI, like lovastatin, may increase their therapeutic activity. MM is a highly aggressive tumor that is rarely curative and median survival is in the range of 10�C17 months, therefore, novel therapies for MM are needed. Elevated levels of circulating and serousal VEGF in MM patients and the expression of VEGF and VEGFR on MM cells that can drive their proliferation and enhance their survival has led to the evaluation of VEGFR targeted therapies. Bevacizumab, a monoclonal antibody against the VEGF, which is approved for the treatment of colon cancer, in combination with chemotherapy, failed to significantly affect outcome to chemotherapy treatment alone. Various VEGFRTKI employed a single agents also failed to demonstrate clinical utility in MM patients. As like HUVEC, MM cells also depend on VEGFR signaling, we also examined the effect of lovastatin alone and in combination with VEGFR-2 TKI on MM cell viability. Combining 5 mM lovastatin treatments with two VEGFR-2 inhibitors in the H28 and H2052 mesothelioma derived cell lines demonstrated synergistic cytotoxicity through the induction of a potent apoptotic response. These results highlight a novel mechanism regulating VEGFR-2 function and a potential novel therapeutic approach for MM. Inhibition of HMG-CoA reductase has been evaluated as an anti-cancer therapeutic approach owing to its ability to inhibit tumor cell proliferation, induce tumor specific apoptosis and inhibit cell motility and metastasis in several tumor models. A number of Phase I Clinical trials evaluating the efficacy of high doses of lovastatin failed to demonstrate significant antitumor activity.
proliferation, survival and migration. VEGF and VEGFR are also expressed by some tumor cells, like MM, acting in a functional autocrine loop capable of directly stimulating the growth and survival of MM cells. In this study, we have shown lovastatin does indeed inhibit ligand-induced VEGFR-2 activation through inhibition of receptor internalization resulting in diminished AKT activation in HUVEC and MM cells. Lovastatin treatment re-organized the actin cytoskeleton, inhibited proliferation and induced apoptosis of HUVEC at therapeutically relevant doses despite addition of exogenous VEGF. AKT activation, which mediates cell survival, along with its downstream targets S6K1 and 4EBP1 were significantly inhibited by lovastatin treatment. Combining lovastatin with VEGFR-TKIs also induced synergistic cytotoxicity of HUVEC cells. Due to their role in promoting tumor neovascularization, inhibiting the function of VEGF and VEGFR has been the focus of a number of therapeutic approaches. The limited clinical responses associated with these agents have been associated with their ability to promote disease stabilization and rarely induce tumor regression. Thus, agents that can cooperate and enhance the activity of VEGFR-TKI, like lovastatin, may increase their therapeutic activity. MM is a highly aggressive tumor that is rarely curative and median survival is in the range of 10�C17 months, therefore, novel therapies for MM are needed. Elevated levels of circulating and serousal VEGF in MM patients and the expression of VEGF and VEGFR on MM cells that can drive their proliferation and enhance their survival has led to the evaluation of VEGFR targeted therapies. Bevacizumab, a monoclonal antibody against the VEGF, which is approved for the treatment of colon cancer, in combination with chemotherapy, failed to significantly affect outcome to chemotherapy treatment alone. Various VEGFRTKI employed a single agents also failed to demonstrate clinical utility in MM patients. As like HUVEC, MM cells also depend on VEGFR signaling, we also examined the effect of lovastatin alone and in combination with VEGFR-2 TKI on MM cell viability. Combining 5 mM lovastatin treatments with two VEGFR-2 inhibitors in the H28 and H2052 mesothelioma derived cell lines demonstrated synergistic cytotoxicity through the induction of a potent apoptotic response. These results highlight a novel mechanism regulating VEGFR-2 function and a potential novel therapeutic approach for MM. Inhibition of HMG-CoA reductase has been evaluated as an anti-cancer therapeutic approach owing to its ability to inhibit tumor cell proliferation, induce tumor specific apoptosis and inhibit cell motility and metastasis in several tumor models. A number of Phase I Clinical trials evaluating the efficacy of high doses of lovastatin failed to demonstrate significant antitumor activity. levels of stimulation, the sensitivity to AICD is further increased by the nuclear import inhibitory peptide. Thus, chronic activation of isletinfiltrating T and B cells in autoimmune diabetes may favor the AICD-enhancing effect of the nuclear import inhibitor. This hitherto unreported action of cSN50 in a relevant preclinical T1D model adds autoimmune inflammation to the list of conditions in which a nuclear import inhibitor has displayed therapeutic utility. We have previously demonstrated the efficacy of nuclear import inhibitor delivery and its anti-inflammatory and cytoprotective action in acute inflammation models, including lethal challenge with superantigen, staphylococcal enterotoxin B, and lipopolysaccharide, which trigger acute inflammatory lung and liver injury.
levels of stimulation, the sensitivity to AICD is further increased by the nuclear import inhibitory peptide. Thus, chronic activation of isletinfiltrating T and B cells in autoimmune diabetes may favor the AICD-enhancing effect of the nuclear import inhibitor. This hitherto unreported action of cSN50 in a relevant preclinical T1D model adds autoimmune inflammation to the list of conditions in which a nuclear import inhibitor has displayed therapeutic utility. We have previously demonstrated the efficacy of nuclear import inhibitor delivery and its anti-inflammatory and cytoprotective action in acute inflammation models, including lethal challenge with superantigen, staphylococcal enterotoxin B, and lipopolysaccharide, which trigger acute inflammatory lung and liver injury. a significantly higher percentage of metaphase arrested cells when treated with ICRF193 and cytospun onto slides to retain all cells.After 18 hour treatments with 2 or 10 mM ICRF-193, or 4 hours with 10 mM ICRF-193, cells with reduced Metnase showed 4.9-fold, 2.2-fold, and 2.6-fold increased metaphase arrest, respectively, as compared to vector control and evaluated by student’s t-test. This result suggests that Metnase promotes decatenation in ICRF-193-treated MDA-MB-231 cells, allowing them to proceed through metaphase even in the presence of this Topo IIa specific inhibitor. Prior studies revealed that bladder and lung cancer cells progress through the decatenation checkpoints when Topo IIa is inhibited by high concentrations of ICRF-193. The conclusion from those studies was that these cancer cells failed to arrest because they had inactivated the decatenation checkpoints. While the ability to progress through mitosis even when Topo IIa is inhibited may be a general feature of malignancy, it may be due to the presence of Metnase alone, or Metnase in combination with checkpoint inactivation. Thus, the decatenation checkpoint may be intact in these malignant cells, but Metnase promotes continued Topo IIa function despite the presence of inhibitors, and the decatenation checkpoint is not activated. The Topo IIa inhibitor ICRF-193 does not induce significant DNA damage, and therefore is not relevant in the clinical therapy of breast cancer. To determine whether altering Metnase levels would affect resistance to clinically relevant Topo IIa inhibitors, such as VP-16 and adriamycin, we determined the cytotoxicity of these agents in MDA-MB-231 cell lines that stably under-expressed Metnase using colony formation assays. Decreased Metnase expression increased sensitivity 7.5-fold to VP-16, and 3.5-fold to adriamycin. Together, these results indicate that Metnase expression levels directly correlate with cell survival after exposure to these clinically relevant Topo IIa inhibitors. Adriamycin is an important agent in both adjuvant therapy and in the treatment of metastatic breast adenocarcinoma, so this finding is of relevance for current clinical
a significantly higher percentage of metaphase arrested cells when treated with ICRF193 and cytospun onto slides to retain all cells.After 18 hour treatments with 2 or 10 mM ICRF-193, or 4 hours with 10 mM ICRF-193, cells with reduced Metnase showed 4.9-fold, 2.2-fold, and 2.6-fold increased metaphase arrest, respectively, as compared to vector control and evaluated by student’s t-test. This result suggests that Metnase promotes decatenation in ICRF-193-treated MDA-MB-231 cells, allowing them to proceed through metaphase even in the presence of this Topo IIa specific inhibitor. Prior studies revealed that bladder and lung cancer cells progress through the decatenation checkpoints when Topo IIa is inhibited by high concentrations of ICRF-193. The conclusion from those studies was that these cancer cells failed to arrest because they had inactivated the decatenation checkpoints. While the ability to progress through mitosis even when Topo IIa is inhibited may be a general feature of malignancy, it may be due to the presence of Metnase alone, or Metnase in combination with checkpoint inactivation. Thus, the decatenation checkpoint may be intact in these malignant cells, but Metnase promotes continued Topo IIa function despite the presence of inhibitors, and the decatenation checkpoint is not activated. The Topo IIa inhibitor ICRF-193 does not induce significant DNA damage, and therefore is not relevant in the clinical therapy of breast cancer. To determine whether altering Metnase levels would affect resistance to clinically relevant Topo IIa inhibitors, such as VP-16 and adriamycin, we determined the cytotoxicity of these agents in MDA-MB-231 cell lines that stably under-expressed Metnase using colony formation assays. Decreased Metnase expression increased sensitivity 7.5-fold to VP-16, and 3.5-fold to adriamycin. Together, these results indicate that Metnase expression levels directly correlate with cell survival after exposure to these clinically relevant Topo IIa inhibitors. Adriamycin is an important agent in both adjuvant therapy and in the treatment of metastatic breast adenocarcinoma, so this finding is of relevance for current clinical  present study set out to develop a rational cell-based drug discovery strategy, an approach that has historically been compromised by the lack of appropriate control cells. With the objective of identifying lead compounds that specifically kill cells with activated Akt signaling and that spare control cells, we have combined the use of co-cultured isogenic cell lines with fluorescent technology. We introduced a myristoylated form of Akt which constitutively localizes to the plasma membrane, bypassing the requirement for PIP3 in Akt activation. This myr-Akt has been shown to constitutively inactivate proapoptotic downstream targets. In order to generate Ba/F3 cells that survive in the absence of IL-3 independent of activated PI3K/Akt signaling.
present study set out to develop a rational cell-based drug discovery strategy, an approach that has historically been compromised by the lack of appropriate control cells. With the objective of identifying lead compounds that specifically kill cells with activated Akt signaling and that spare control cells, we have combined the use of co-cultured isogenic cell lines with fluorescent technology. We introduced a myristoylated form of Akt which constitutively localizes to the plasma membrane, bypassing the requirement for PIP3 in Akt activation. This myr-Akt has been shown to constitutively inactivate proapoptotic downstream targets. In order to generate Ba/F3 cells that survive in the absence of IL-3 independent of activated PI3K/Akt signaling.