Ghrelin is a 28-amino acid peptide, produced by the stomach and hypothalamic neurons. It is the endogenous ligand of the growth hormone secretagogue receptor 1a. Ghrelin receptors are expressed by various brain regions, such as the arcuate nucleus, lateral hypothalamus, VMH and suprachiasmatic nucleus, Evofosfamide inquirer structures known to be involved in feeding and sleep regulation. Ghrelin secretion is stimulated by fasting and ghrelin enhances feeding and increases adiposity in rats. Growing body of evidence indicates that ghrelin signaling plays a role in the function of arousal mechanisms. Systemic, intracerebroventricular or intrahypothalamic administration of ghrelin suppresses sleep in rats. Ghrelin receptor KO mice show attenuated arousal responses to food deprivation and to the exposure of novel  environment. Ghrelin is also implicated in the function of thermoregulatory mechanisms and in the integration of sleep and thermoregulatory responses. Central administration of ghrelin diminishes the activity of brown adipose tissue, a key effector organ in non-shivering thermogenesis, by suppressing the activity of its sympathetic innervation. The product of the preproghrelin gene play a role in coordinating thermoregulatory/metabolic and sleep responses to metabolic challenges. When fasted in the cold, normal mice develop hypothermic bouts and increased sleep during these hypothermic periods. Ghrelin deficient preproghrelin knockout mice are incapable of mounting sleep responses under these conditions and enter precipitous, lethal, hypothermia. FAS inhibitors, such as C75 greatly suppress ghrelin production by the stomach and the hypothalamus. C75 potently suppresses eating and energy expenditure. Since ghrelin stimulates feeding and transgenic mice with elevated circulating ghrelin levels have increased energy expenditure, it seemed possible that the inhibitory effects of C75 on feeding and energy expenditure are mediated by its suppressive action on ghrelin production. To test this hypothesis, we determined the effects of C75 on feeding, metabolism, sleep and motor activity in ghrelin receptor deficient mice. Our major finding is that systemic injection of C75 suppresses motor activity, REMS, and SWA of the EEG in both normal and ghrelin receptor KO mice. These behavioral and sleep effects are accompanied by decreases in VO2, body temperature and RER. We confirm our and others’ previous findings that spontaneous sleep-wake activity, motor activity and food intake on standard laboratory diet are not affected in ghrelin receptor KO mice. Our results also confirm that C75 elicits robust dose-dependent inhibition of 24-h food intake. The effects of C75 on the daily rhythm of feeding have not been reported before. We show that C75 abolished the diurnal rhythm of feeding. Night-time food intake was decreased to the level normally seen during the day, the rest period in mice. The effects of C75 on motor activity have not been quantified previously. In two studies, no “gross Nilotinib changes” in the activity of mice or no “obvious motoric effects” in rats were observed after systemic injection of C75. In the present study, we quantified the effects of C75 on spontaneous motor activity in two separate sets of experiments, by using two different methods. We found that spontaneous motor activity was decreased after ip injection of C75 as measured both by the movement of implanted transponders over a horizontal receiver in Experiment 1 and by the interruption of infrared beams in Experiment 2. The mechanism of the anorectic effects of C75 is not well understood. It is possible that C75 stimulates satiety circuits and/ or suppresses orexigenic mechanisms or inhibits feeding by eliciting visceral illness.
environment. Ghrelin is also implicated in the function of thermoregulatory mechanisms and in the integration of sleep and thermoregulatory responses. Central administration of ghrelin diminishes the activity of brown adipose tissue, a key effector organ in non-shivering thermogenesis, by suppressing the activity of its sympathetic innervation. The product of the preproghrelin gene play a role in coordinating thermoregulatory/metabolic and sleep responses to metabolic challenges. When fasted in the cold, normal mice develop hypothermic bouts and increased sleep during these hypothermic periods. Ghrelin deficient preproghrelin knockout mice are incapable of mounting sleep responses under these conditions and enter precipitous, lethal, hypothermia. FAS inhibitors, such as C75 greatly suppress ghrelin production by the stomach and the hypothalamus. C75 potently suppresses eating and energy expenditure. Since ghrelin stimulates feeding and transgenic mice with elevated circulating ghrelin levels have increased energy expenditure, it seemed possible that the inhibitory effects of C75 on feeding and energy expenditure are mediated by its suppressive action on ghrelin production. To test this hypothesis, we determined the effects of C75 on feeding, metabolism, sleep and motor activity in ghrelin receptor deficient mice. Our major finding is that systemic injection of C75 suppresses motor activity, REMS, and SWA of the EEG in both normal and ghrelin receptor KO mice. These behavioral and sleep effects are accompanied by decreases in VO2, body temperature and RER. We confirm our and others’ previous findings that spontaneous sleep-wake activity, motor activity and food intake on standard laboratory diet are not affected in ghrelin receptor KO mice. Our results also confirm that C75 elicits robust dose-dependent inhibition of 24-h food intake. The effects of C75 on the daily rhythm of feeding have not been reported before. We show that C75 abolished the diurnal rhythm of feeding. Night-time food intake was decreased to the level normally seen during the day, the rest period in mice. The effects of C75 on motor activity have not been quantified previously. In two studies, no “gross Nilotinib changes” in the activity of mice or no “obvious motoric effects” in rats were observed after systemic injection of C75. In the present study, we quantified the effects of C75 on spontaneous motor activity in two separate sets of experiments, by using two different methods. We found that spontaneous motor activity was decreased after ip injection of C75 as measured both by the movement of implanted transponders over a horizontal receiver in Experiment 1 and by the interruption of infrared beams in Experiment 2. The mechanism of the anorectic effects of C75 is not well understood. It is possible that C75 stimulates satiety circuits and/ or suppresses orexigenic mechanisms or inhibits feeding by eliciting visceral illness.
The role of orexinergic mechanisms in the effects of C75 to play a role in arousal responses to fasting
Leave a reply
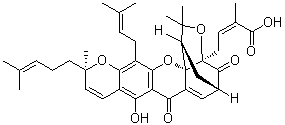 receptor KO mice indicates that these effects of C75 are unrelated to the function of ghrelin receptors. We found significant correlation between the anorectic and motor activity effects of C75. An overall decrease in motor activity may, in theory, lead to decreased feeding. Although causality cannot be determined with certainty in the present study, it seems unlikely that the anorectic effects are exclusively due to the general
receptor KO mice indicates that these effects of C75 are unrelated to the function of ghrelin receptors. We found significant correlation between the anorectic and motor activity effects of C75. An overall decrease in motor activity may, in theory, lead to decreased feeding. Although causality cannot be determined with certainty in the present study, it seems unlikely that the anorectic effects are exclusively due to the general 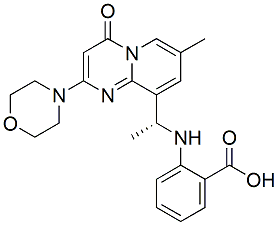 structureactivity relationship analyses. In fact, several structural analogues of these compounds exist such as secomanoalide and luffariellolide for manoalide and L538,916 for MK-886, thus enabling the initiation of such studies. In summary, we presented herein the development of new strategies for the discovery of small molecules that could inhibit pol k activity both in vitro and in vivo. The identification of chemotypes with established drug properties targeting pol k validates this qHTS platform, as well as the secondary assays and sets the stage for exploration of significantly
structureactivity relationship analyses. In fact, several structural analogues of these compounds exist such as secomanoalide and luffariellolide for manoalide and L538,916 for MK-886, thus enabling the initiation of such studies. In summary, we presented herein the development of new strategies for the discovery of small molecules that could inhibit pol k activity both in vitro and in vivo. The identification of chemotypes with established drug properties targeting pol k validates this qHTS platform, as well as the secondary assays and sets the stage for exploration of significantly 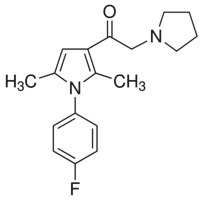 slow exchange, indicative of a Kd likely in the low mM range. In contrast, when performed in the presence of saturating ATP or AMPCPP, widespread perturbations were observed in slow exchange for all resonances, despite no change in binding affinity measured by ITC and SPR. Chemical shift perturbations clearly mapped to the respective substrate and cofactor site. This work reports the discovery, binding properties and mechanism of a novel, competitive pterin site inhibitor, presented in complex with the first crystal structure of SaHPPK. The pterin site is highly specific and restricts the chemical space available for inhibitor design to structures closely resembling the pterin scaffold. Consequently, the literature is devoid of non-pterin like HPPK inhibitors, despite mounting structural information that has been reported over the last decade. In line with the high pterin-site specificity is the high ligand efficiency of 8-mercaptoguanine. 8-Mercaptoguanine has previously been reported to have biological activity. Early studies revealed some lipolytic
slow exchange, indicative of a Kd likely in the low mM range. In contrast, when performed in the presence of saturating ATP or AMPCPP, widespread perturbations were observed in slow exchange for all resonances, despite no change in binding affinity measured by ITC and SPR. Chemical shift perturbations clearly mapped to the respective substrate and cofactor site. This work reports the discovery, binding properties and mechanism of a novel, competitive pterin site inhibitor, presented in complex with the first crystal structure of SaHPPK. The pterin site is highly specific and restricts the chemical space available for inhibitor design to structures closely resembling the pterin scaffold. Consequently, the literature is devoid of non-pterin like HPPK inhibitors, despite mounting structural information that has been reported over the last decade. In line with the high pterin-site specificity is the high ligand efficiency of 8-mercaptoguanine. 8-Mercaptoguanine has previously been reported to have biological activity. Early studies revealed some lipolytic 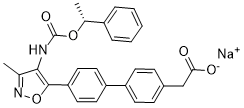 beyond a threshold of 10 MPK, most of this compound is rapidly removed by excretion and therefore further reduction in tumor volume was not observed. Previously, D-PDMP has been used extensively to examine the role of glycosphingolipid and related glycosytransferases in arterial smooth muscle cell proliferation, wound healing, osteoclastogenesis,
beyond a threshold of 10 MPK, most of this compound is rapidly removed by excretion and therefore further reduction in tumor volume was not observed. Previously, D-PDMP has been used extensively to examine the role of glycosphingolipid and related glycosytransferases in arterial smooth muscle cell proliferation, wound healing, osteoclastogenesis,